The picture below shows the different stages of regulatory control.
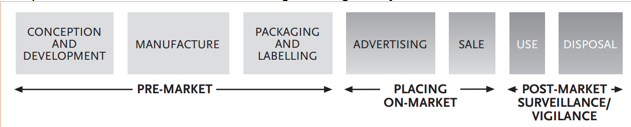
Post-Market Surveillance is the phase in which the product is placed on the market. This also requires several requirements for the manufacturer that are described in this section.
After the initial Clinical Evaluation (see section Verification and Validation) clinical follow up is needed because the product is now on the market and actually used on patients. Periodically new clinical data from the market is gathered via new literature assessment, patient case reports, complaints, etc). If needed a Post Market Clinical Follow up (PMCF) is executed. See MEDDEV 2.12/2, Post Market Clinical Follow-up studies for guidance.
The Technical File including the Risk Management File (see section Risk Management) must be kept up-to-date with feedback from post market data.
Customer complaints must be analysed and followed up. The following question needs to be answered for the occurred event recorded in a complaint:
Has the medical device caused or contributed to a death or unanticipated serious deterioration in the state of health of a patient, user or other person or would it do if the event were to recur?
If above is answered with “yes”, the event is considered a serious incident and a report must be sent to the Competent Authorities and Notified Body. This is known as the Vigilance procedure. A strict reporting timing constraint is defined: within 10 days after becoming aware of the serious incident. See MEDDEV 2.12/1 Medical Devices Vigilance System for guidance on vigilance.